Niemann-Pick C (NPC) is a tragic neurodegenerative disease that is often referred to as childhood Alzheimer’s disease. 95% of patients with NPC carry mutations in the npc1 gene, which codes for the Npc1 protein. Npc1 resides in lysosomes and is necessary for cholesterol export out of the lysosome. Cells with a defective npc1 gene accumulate lysosomal cholesterol, among other defects. Humans, cats, and mice with npc1 mutations lose control of bodily movements due to neurodegeneration of the Purkinje neurons (PKs) of the cerebellum.
In the NPC research field, it is debated why PKs are hypersensitive to the loss of Npc1 function. Here are four possible explanations (see figure below). (1) Npc1 protein is required for proper export of cholesterol from lysosomes (grey spheres). Perhaps the accumulation of cholesterol in NPC is itself toxic to lysosomes, and PKs have an elevated need for lysosomal function. (2) The sequestration of cholesterol in lysosomes prevents it from reaching other intracellular destinations, where it would ordinarily be used for normal/healthy cellular processes. PKs may require a higher concentration of cholesterol for their viability. (3) Other scientists have observed death of glia in NPC mice. Perhaps glia or other nearby cells (green cells) secrete an important signal that maintains the viability of PKs, and that signal is not secreted when npc1 is mutant. (4) Death of glia or other cells leads to toxic elements entering the cellular environment and these lead to PK death (all figures here are from Ko, et. al, 2005).

Note that by definition, the death of PK neurons in models 1 and 2 originates from problems within a soon-to-die PK. In contrast, models 3 and 4 necessitate that the root cause of PK death originates from outside PKs. Luckily, crafty experiments can be done in mice to test whether the origin of the detrimental events arise from inside or outside the PKs.
In 2005, Stanford scientists created mice with some wild type and some npc1 mutant cells. That was done by mixing early stage embryos from wild type and npc1 mutant mice. Seven mosaic mice, ranging from 40% to 90% mutant for npc1, were selected for analysis. If model 3 is true, then the death of npc1 mutant PKs will not be as severe if they are surrounded by a sea of wild type cells. If model 4 is true, then wild type PKs will die when surrounded by npc1 mutant cells. If neither models 3 or 4 are true, then models 1 and 2 become favored.
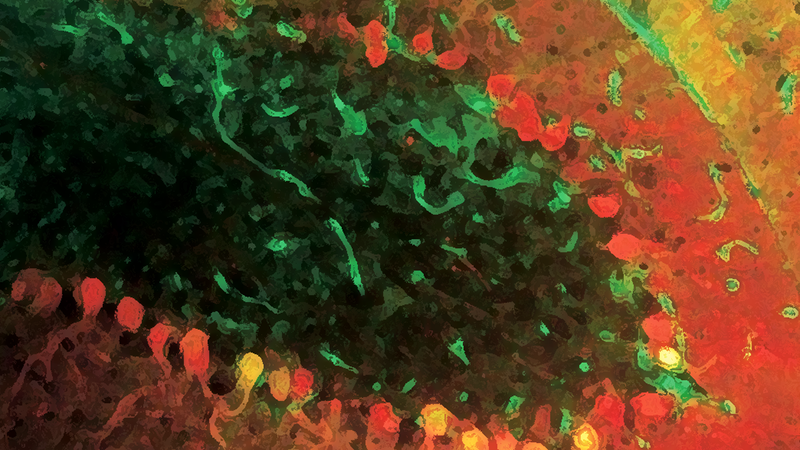
I’ll cut to the chase and tell you that they ruled out models 3 and 4, and therefore found that Npc1 is required to act within PK neurons to prevent their death. The figures below are sections of a cerebellum from a 70 day old mouse that was 33% wild type and 66% npc1 mutant. The upper panel is cerebellar lobule X. Lobule X is unique in that it’s resistant to PK death in npc1 mutant mice, and looks wild type in 70 day old animals. As expected, there are several red staining PKs in lobule X. The lower panel is cerebellar lobule II, a region that normally exhibits a high degree of PK death by day 70 in npc1 mutant mice. Despite this animal being 33% wild type, there is still a high degree of PK death in lobule II as if the entire tissue were npc1 mutant. This trend held in their other six mice, and as much as 95% of their PKs mutant for npc1 died. These data rule out Model #3, which poses that in wild type animals, a hypothetical signal is produced from surrounding cells that promotes PK neuron viability. Furthermore, in Model #3, npc1 mutant animals have a diminished pro-viability signal because the cells that produce it die. Since lots of wild type cells are still present in the mouse cerebella depicted below, npc1 mutant PK cells should have stayed alive if Model 3 were true. They also found that wild type PK cells did not die despite the presence of patches of npc1 mutant tissue (not shown). That observation rules out Model #4, where a “death” signal originates from dying npc1 mutant cells.

Knowing that the detrimental element must originate from within the soon-to-die PK cells, Models #1 and #2 look favorable. Both of those models put forth that cholesterol accumulation in PKs leads to their death. As noted above, lobule X of the cerebellum is resistant to PK degeneration.
Below are the authors’ observations of the level of cholesterol accumulation (filipin dye) in a wild type lobule (left), npc1 lobule II (middle), and npc1 lobule X (right). The degree of cholesterol accumulation in lobule X is the same as that of the other lobules. So, if cholesterol accumulation is toxic, why don’t the PKs in lobule X degenerate? This implies that cholesterol accumulation itself is insufficient to cause PK death. However, I’m not fully satisfied with the use of lobule X as a control for that experiment. Since it’s resistant to PK death in NPC, its PKs may have some unique feature protecting them from neurodegeneration in NPC.

Survival of lobule X PKs despite cholesterol accumulation in NPC is intriguing, and should be explored further. It suggests that cholesterol accumulation to possibly toxic levels in lysosomes is insufficient to cause PK death. That interpretation would rule out Model #1. However, it’s likely that most of the cholesterol is trapped in lysosomes, preventing it from being utilized by the cell (Model #2). Thus, the similar amount of cholesterol accumulation in the resistant PK neurons of Lobule X versus the other, sensitive, lobules could be interpreted to rule out Models 1 and 2. Hmmm..the debate continues. I think Models 1 and/or 2 to explain PK death in NPC should still be the favored, “placeholder,” models until one of them or a new model is proven. What we can safely conclude from this study is that something goes awry within PKs in NPC, and this intrinsic problem leads to their death. Thus, an NPC therapy meant to correct PK neurodegeneration needs to be delivered to, and functional within, PKs.
