The Condition: Mucolipidosis Type IV
Mucolipidosis is a group of inherited lysosomal storage disorders characterized by aberrant storage and accumulation of lipid vesicles in patient cells. Mucolipidosis type IV (MLIV) is a rare genetic lysosomal storage disorder that presents itself in the first year of life and is characterized by neural as well as ophthalmologic abnormalities. Ophthalmologic abnormalities include corneal clouding and strabismus (misalignment of the eyes). These abnormalities can be observed right after birth and are progressive in nature even though the disease is known to stabilize during the second and third decades of the life of a patient. In patient cells, there is an abnormal accumulation of phospholipids and mucopolysaccharides. Mutations in the MLIV gene, mucolipin 1 (MCOLN1), are the cause of this abnormal mucolipin) and has six transmembrane domains. TRPML1 is a cation permeable channel which may play an important role in mediating the release of Ca2+, Fe2+ and Zn2+ into late endosomes or lysosomes. The function of TRPML1 is not completely understood but is thought to play an important role in trafficking events and ionic homeostasis in the cell.
Using Perlara Platform for Screenotype
Currently there are no treatments for this disease, so we are using the Perlara platform to identify potential therapeutics. In order to do this, we optimized a patient cell based phenotypic screen to discover compounds that rescue the disease. To get our screenotype, we started off with our assay development phase. The first step was to obtain MLIV fibroblast cell lines. We obtained four MLIV patient fibroblast cell lines from the Coriell Institute cell repository. While expanding and freezing down these cell lines, we found that the GM02527 B line was a very robust and fast-growing cell line in our growth conditions. This cell line also has the most common mutation observed in patients, making up 72% of MLIV patient population. Therefore, we went ahead and conducted all of our phenotyping on this particular cell line in 96 well black walled glass bottomed plates. All throughout this post, when I mention MLIV cells, I would be referring to GM02527B cells. Based on our literature review, we shortlisted a few readouts to test; LysoTracker (lysosome accumulation), FluoZin (Zinc accumulation), LC3 (autophagy), P62 (autophagy), Filipin (cholesterol accumulation) and Bodipy LacCer (sphingolipid accumulation).
MLIV Screen Evaluation
In order to check whether MLIV fibroblasts accumulate free cholesterol like the first lysosomal storage disease we studied, Neimann Pick Type C, we performed the filipin stain assay (described here in a previous post by Nina). The results we obtained let us conclude that filipin fluorescence / cholesterol accumulation was not intense enough in MLIV cells for a large-scale screen. Another thing that is known to be seen in MLIV cells is zinc dyshomeostasis, which manifests as an accumulation of chelatable Zn2+ in the lysosomes of cells. We did a couple of experiments to narrow down the phenotype of Zn2+ accumulation in lysosomes with a colocalization experiment using FluoZin-3™ (membrane-permeable fluorescent dye) and LysoTracker dyes. The FluoZin-3™ signal was not intense and consistent enough for a screen. Therefore, we decided not to move forward with this phenotype either. Another dye that we tested was Bodipy LacCer to check whether MLIV fibroblasts had a phenotype of accumulating lactosylceramide (LacCer). We tested Bodipy LacCer dye in various media plus serum conditions and found that there was no difference between signals in MLIV and WT cells. Therefore, we quickly ruled out using filipin, FluoZin-3™, and Bodipy LacCer as readouts for an MLIV screen.
LysoTracker Phenotype
We know that MLIV is a lysosomal storage disorder, and we wanted to check whether we observe an accumulation of lysosomes. Hence, we tested this phenotype using the LysoTracker dye and found that there is a significant difference between the signals of MLIV and WT fibroblasts as shown below by the increase in fluorescence in MLIV cells on the right (Figure 1). In the literature, LysoTracker stained cells were always imaged live and since we were still setting up our live cell imaging scope, we tested a number of time-points and imaged both live and fixed cells. Below is an example image of cells stained with lysotracker for one hour, then fixed and imaged. Since, there is such a big difference in the signals between the MLIV and WT cells (Figure 1), the LysoTracker phenotype is a promising screenotype.

Figure 1. Images representing lysosome accumulation phenotype that shows the huge difference between LysoTracker staining in WT and MLIV cells (20x images)
Autophagic Dysfunction
Autophagic dysfunction is another phenotype that has been reported in MLIV cells. Autophagy is a process by which cells degrade proteins and cell organelles in bulk. The process of autophagy begins when an autophagosome is formed by the sequestration of a portion of the cytoplasm by a double membrane structure that grows and seals off. We chose LC3 because it is one of the most selective markers for autophagosomes. An increase in LC3 would correlate to either an increase in autophagy or an accumulation of autophagosomes because of a block in autophagy. As the process of autophagy moves along, autophagosomes may fuse with endosomal multivesicular bodies (MVB) or lysosomes. Autophagosomes fuse with lysosomes to form autolysosomes allowing the contents of autophagosomes to be degraded by lysosomal enzymes. As I mentioned earlier, MCOLN1 is responsible for the abnormal accumulation of proteins and lipids. This suggests that MCOLN1 plays an important role in the correct transferring/ moving of proteins and lipids in the late endosomal/lysosomal pathway. The absence of MCOLN1 was seen to increase basal autophagy levels and as a result decreased autophagosome degradation (Silvia Vergarajauregui, 2008). An accumulation of P62 (suggesting an abnormal accumulation of ubiquitinated proteins) is also seen in MLIV patient fibroblasts as compared to normal fibroblasts (Silvia Vergarajauregui, 2008).
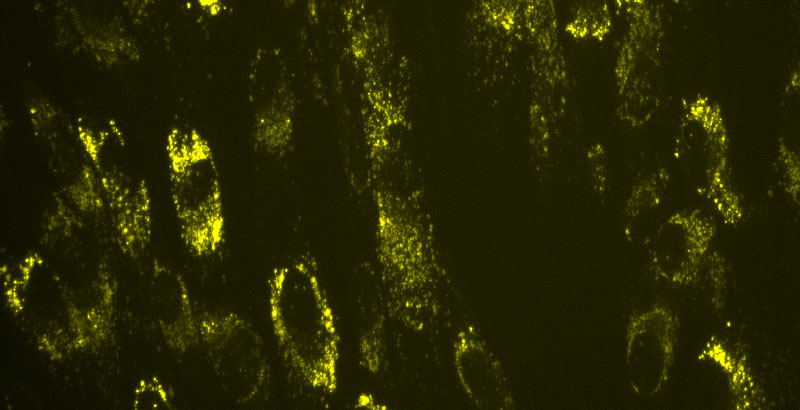
Autophagy is upregulated when fibroblasts are starved and can be used to enhance the LC3 and P62 accumulation phenotype. We starved the fibroblasts in Earle’s balanced salt solution (EBSS) for 3 hours (Figure 2) and then fixed, blocked, stained (primary and secondary antibodies) and imaged. We observed a slight increase in accumulation of LC3 in MLIV fibroblasts as compared to normal WT fibroblasts. This is observed because MLIV cells have a block in autophagy and by inducing autophagy with starvation, you are increasing the accumulation. An even greater difference is observed in the accumulation of P62 in MLIV versus WT cells (Figure 2 bottom right panels).

Figure 2. Images representing the enhanced LC3/ P62 phenotype by starvation in EBSS for 3 hours. Images also represent the increased accumulation of LC3 as well as P62 staining in MLIV cells as compared to WT cells. (40x images)
Screen design
The only phenotype that shows great promise and ease in scaling up is the lysosomal accumulation. Before we scaled up, we obtained and tested a few control compounds on MLIV cells to see whether we could reverse the phenotype. Of the four compounds we tested, MK6-83, a TRPML1 channel activator, was found to reverse MLIV LysoTracker phenotype at 20 µM (Figure 3). We were also able to confirm the same using our MetaXpress Custom Module Editor for quantifying the lysosomal puncta area.

Figure 3. Images representing the MLIV lysosomal accumulation reversal phenotype with MK6-83 at 20 µM. (40x images)
Secondary Screen
LC3/ P62 was another assay that we could use for a screen but none of the control compounds worked to reverse the accumulation phenotype in the starvation conditions tested. To proceed with a screen, we would have to use WT as a control on all the plates. Also, other points to be considered, in doing a large-scale screen with this assay would be the long starvation time, and the long immunostaining protocol to be followed before these plates can be imaged. Thus, we decided to keep this assay to be used as a secondary screen.
We are in the process of scaling up these assays from 96 to 384 well plates and conducting a screen. We are looking forward to finding hits and comparing these hits to the ones from our Drosophila and C. Elegans models. Keep a lookout for our next MLIV blog post for an update!
References
Silvia Vergarajauregui, P. S. (2008). Autophagic dysfunction in mucolipidosis type IV patients. Human Molecular Genetics, 2723-2737.
