Last month I traveled to Tucson for my third consecutive Ara Parseghian Medical Research Foundation meeting for Niemann-Pick Type C/NPC. In 2014 at my first APMRF meeting, which was hosted in South Bend at Notre Dame University, I was a newcomer to the NPC community but eager to introduce PLab’s platform to the academic brain trust and committed families who annually gather for this conference/reunion.
![]()
The ink on PLab’s Articles of Incorporation was barely dry. Our core team of scientists – and the lab itself – was just assembling in San Francisco. A year later, I returned to South Bend along with PLab Senior Scientists Nina and Tom to announce the completion of a two-invertebrate (worm & fly), 50,000-compound NPC high-throughput (HTS) screening campaign, and the discovery of a novel compound that we named PERL101. As I’ll describe below, all we knew at that time was that PERL101 achieved two-factor authentication: (1) PERL101 rescues NPC1 mutant worm larvae to adulthood and (2) PERL101 modifies (in an unexpected manner) the diagnostic cellular phenotype of NPC disease in immortalized patient-derived fibroblasts.
This year’s APMRF returned home to Arizona after a sojourn in the Midwest. I presented a poster chronicling the progress of PERL101 from HTS hit compound in a nematode development phenotypic screen in May 2015 to lead compound in a NPC1 knockout (KO) mouse efficacy study in July 2016– without any intervening chemical optimization. In Part 1 and Part 2 of this research-in-real-time series, I shared these data. Here I share additional data on PERL101, including a multi-dose PK study in wild-type mice, a pilot MTD study in NPC1 KO mice, and preliminary mechanism-of-action (MOA) experiments in NPC patient fibroblasts.
I left off in Part 2 with a summary of a 90-day maximum tolerated dose (MTD) study with PERL101 on young adult wild-type Balb/c male mice. We identified a MTD of 80 mg/kg (mpk). We observed an unusual dose response from 80 mpk to 60 mpk with respect to body weight change over time; 60 mpk looks like the inverse of 80 mpk, with the former exhibiting decelerated body weight gain and the latter exhibiting accelerated body weight gain. We also noted that body weight changes manifested only after chronic (many week) dosing. This result suggested to us that PERL101, while well tolerated in wild-type mice, might exhibit non-trivial tissue accumulation over time. (We’re in the process of having the MTD histopath slides archived by our YC batchmate Histowiz; more on that in the next installment).
To gain deeper insight into PERL101’s pharmacokinetics (PK), we measured the concentration of PERL101 in plasma and across a panel of tissues, namely liver, spleen, kidney and lung, over seven days of oral daily dosing at 80 mpk, the MTD in wild-type mice, followed by a 7-day washout to assess clearance rate. Below is a plot of PERL101 concentration in plasma and liver over the 2-week study (the washout period begins at the dashed line):

Several observations are noteworthy. First, PERL101 accumulation is saturable. In liver and plasma, PERL101 peak levels are the same after two daily doses as after seven daily doses. In other words, for the pharmacokineticists in the audience, Cmax is unchanged over a week of continuous daily dosing. Second, PERL101 accumulates in liver at least hundred-fold more than in plasma. Third, PERL101 rapidly equilibrates and rapidly clears. It only takes 48 hours for PERL101 levels to peak in both liver and plasma. Once daily dosing was discontinued after seven days, clearance of PERL101 from liver and plasma essentially followed nearly identical kinetics as accumulation. PERL101 binding is reversible and it rapidly diffuses from sites of accumulation. The above plot is log-scale so values of zero cannot be plotted, but after seven days of washout PERL101 was in fact undetectable in liver, while trace amounts persisted in plasma.
Next we compared the tissue accumulation data from the multi-dose PK study to the tissue accumulation data from the 90-day MTD study. As is apparent in the above figure, the liver-to-plasma ratio of PERL101 is extraordinarily high after only two days of oral dosing. What happens to PERL101 after 90 days of continuous daily dosing? Below is a bar plot of PERL101 concentration in liver after two days, after seven days and after ninety days of daily oral exposure:

Remarkably, PERL101 achieves the same level of accumulation in liver after the second dose, after the seventh dose and after the ninetieth dose. In other words, Cmax is constant. As far as desirable pharmaceutical properties go, this result is encouraging. It means PERL101 Cmax can be predictably controlled by dose, and that PERL101 is rapidly cleared from liver (and presumably the body, though that still has to be established on a tissue-by-tissue basis) after cessation of treatment.
With those data in hand, we performed a pilot tolerability study with a handful of NPC1 KO mouse to assess whether the MTD in wildtype mice is also the MTD in Day 35 KO mice, the treatment paradigm intervention point of our definitive efficacy study. At Day 35, NPC1 KO mice are asymptomatic with respect to CNS disease but have advanced disease in the periphery, meaning liver, spleen, lung, etc. Here’s a plot of body weight of three animals dosed with 80 mpk and four animals dosed with 60 mpk, along with an (imperfect) control group of vehicle-treated KO animals from our cyclodextrin replication study:

One result is obvious: Day 35 NPC1 KO mice do not tolerate PERL101 at 80 mpk as well as wild-type mice. (Mice were weighed until they dropped more than 20% of their peak body weight). Given the prodigious accumulation of PERL101 in the liver and the deterioration of liver drug-metabolism function in Day 35 animals (as evinced by elevated ALT and AST levels and previously published results here), the toxicity of PERL101 at 80 mpk is not entirely surprising.
Encouragingly, even though 80 mpk proved to be too toxic a dose, the 60 mpk cohort (again, please note that n=4) look essentially like vehicle-treated animals, though one individual did maintain weight beyond Day 65, (which we never observed in the vehicle-treated group). Whether or not the 24-hour moving average of weight is different in the 60 mpk group compared to the vehicle-treated control group, these results indicated to us that any efficacy study would have to be performed with lower doses of PERL101 so that toxicity would not obscure any potential efficacy.
In fact, PERL101 accumulates to millimolar concentrations in kidney and spleen as well, as shown in this plot summarizing data from the wildtype MTD study:

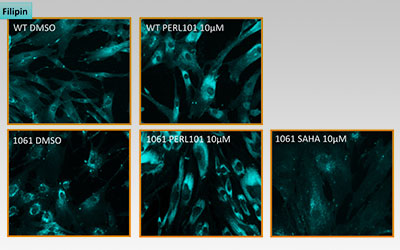
I’ll conclude with some MOA data. Out of 50,000 compounds screened, 100 worm hits emerged after multiple rounds of rigorous retesting. All 100 were tested on NPC patient fibroblasts in the filipin assay, which stains intracellular free cholesterol. We tested fibroblasts from a NPC patient homozygous for I1061T, the most common missense mutation. 14 out of 100 hits were active in NPC patient fibroblasts, but not how we anticipated. In fact, PERL101 was the first hit out of those 14 that displayed a cholesterol phenotype that was the opposite of what we expected based on the NPC literature.
The figure below shows the result of a filipin staining assay with wildtype (WT) and NPC patient fibroblasts (1061) after 24 hours of drug or vehicle treatment:

Three things stand out. First, PERL101 induces an unexpected redistribution and modest increase (confirmed by GC-MS) of intracellular free cholesterol; this effect is seen in both normal and diseased cells. Second, PERL101’s redistribution effects are the opposite of SAHA’s clearance effects; SAHA is a proteostatic modulator and rescues folding defects of NPC1 missense mutations like I1061T. Third, the PERL101-induced filipin phenotype is not an exaggerated or amplified version of the basal NPC disease phenotype.
We were expecting that PERL101 would clear the cholesterol storage phenotype but instead our worm HTS revealed a third way out, a pharmacological bypass suppressor. These results square quite nicely with the original rescue of NPC1 mutant worm development. Diseased larvae don’t progress to adulthood because they can’t produce dafachronic acid, the cholesterol-derived hormone that drives worm development. In short, PERL101 makes cholesterol bioavailable again in both worms and patient fibroblasts.
If we combine the primary worm screen with the secondary fibroblast assay, a concise model of MOA emerges. PERL101 rescues NPC1 mutant larvae to adulthood by making cholesterol more bioavailable through a redistribution and modest increase of free cholesterol. That mean PERL101’s MOA is completely different from cyclodextrin’s or SAHA’s or arimoclomol’s, the three experimental drugs currently in repurposing clinical trials for NPC. And in a nod to whole-animal phenotypic screens, it’s a safe bet that if PERL101 had been identified in a cell-based screen using the filipin assay without any context from a NPC worm primary screen, it would be have likely been tossed out as an enhancer or ignored because it didn’t clear cholesterol storage.
One parting thought. Some of you may be wondering why I omitted MAA data until now. It’s not because we haven’t been exploring MAA this entire time. Quite the contrary, we’ve been using NPC patient fibroblasts and other cells types in a batter of assays and in conjunction with our NPC worm model to understand what PERL101 is doing at the molecular and cellular level. We are committed to Open Science, but we also want to have the highest possible confidence in our data before sharing it on the blog, and ultimately one day (relatively soon) in a peer-reviewed, open-access publication.
Data from our NPC1 KO mouse efficacy study, now underway at Vium, will be streaming in over the coming weeks and months. Next update in early Fall!
