A few months ago, Taryn Sumabat blogged about development of a fly model of a Niemann-Pick Type A Disease. Here I’ll remind you of the progress she described, and update you on our more recent work. If you remember, the two main objectives for this phase of our NPA PerlQuest were to: 1. generate a fly model of NPA by creating null and hypomorphic mutants, and 2. to characterize these mutants to discover phenotypes suitable for a high-throughput screen (“screenotypes”). The fly ortholog of SMPD1 (CG3376 or smpd1) shares ~42% protein identity with human SMPD1. While the function of smpd1 in flies is uncharacterized, it is expressed in structures that will form the pharynx, esophagus, and trachea during embryonic development.
Creating ASM mutants
We first created a null mutant with a large deletion in smpd1. Using a homology directed repair strategy with the CRISPR/Cas9 genome editing system, we deleted and replaced nearly the entire coding sequence of smpd1 with a red fluorescent protein (dsRed) (Figure 1). We found that flies homozygous for this smpd1 null allele are inviable, dying before reaching the adult stage.

Figure 1. A CRISPR/Cas9 strategy to generate a smpd1 null allele using homology directed repair (HDR). Double strand breaks were targeting the first and last exons of the smpd1 locus. A donor plasmid encoding the fluorescent protein dsRED flanked by dSmpd1 homology arms was integrated into the genomic locus.
Next, to generate hypomorphic smpd1 alleles we used a CRISPR/Cas9-based non-homologous end joining strategy. We targeted double strand breaks in the region of smpd1 locus encoding the C-terminal tail of the dSMPD1 protein to generate insertion/deletion events. We reasoned that these indels could produce truncated forms of SMPD1 protein that retained an active catalytic domain. Using this strategy we isolated 15 unique alleles of dSmpd1, including ten protein truncations, three small deletions, and two small insertions (Figure 2). We found, however, that all of these mutations, like the null allele, are also homozygous lethal at the adult stage. Furthermore, we found no combination of these alleles that are viable as compound heterozygotes.
Figure 2. A CRISPR/Cas9-mediated non-homologous end joining strategy to generate smpd1 indel alleles. CRISPR guide RNA were designed to target double strand breaks in a region encoding the C-terminal domain of SMPD1 protein. We isolated 15 unique indel alleles.
Characterizing early death
We next sought to characterize when these smpd1 mutant homozygotes die, first testing the alleles we expected to have the weakest effect on smpd1 function. Specifically, we investigated the alleles that introduced a 24-base pair deletion, or a 9-base pair insertion into the gene. These alleles do not truncate the protein with an early stop, but rather just remove 8 or add 3 amino acids to Smpd1 protein, respectively.
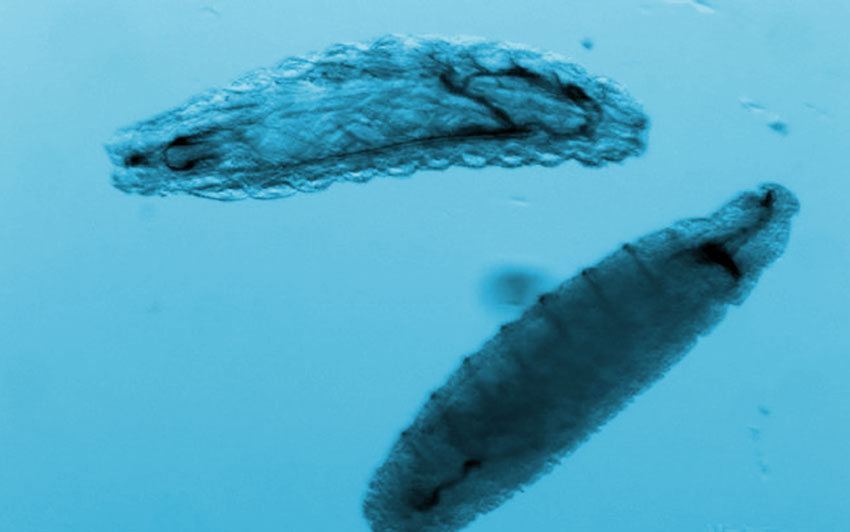
We isolated populations of smpd1 heterozygote and homozygote animals during late embryogenesis as they hatch into larvae (Figure 3). Like the smpd1 homozygous null animals that Taryn described previously in the last NPA update, many 1st instar larvae homozygous mutant for either the 24-bp deletion, or a 9 bp insertion (data not shown) in smpd1 were dead or dying after isolation (Figure 3). These smpd1 mutant homozygotes were small, exhibited abnormal locomotor and feeding movements, and displayed incomplete tracheal development compared to heterozygous larvae carrying a wild-type allele. These phenotypes seem consistent with where smpd1 is expressed during embryo development. Therefore, the homozygous animals appear able to proceed through embryonic development but die soon after hatching. Because these Drosophila mutants died so early in development, we were unable to obtain an adequate tissue sample to measure the acid sphingomyelinase activity and sphingomyelin levels in homozygous mutant flies.
Only 10% of reported patient Smpd1 mutations are in the C-terminal domain, and the majority of these mutations cause Niemann-Pick Type B, the non-neuropathic form of the disease (Zhou et al., 2016). At the extreme, one such mutation is a single amino acid deletion, ∆R608 ( Levran et al., 1991) which could signal that there is little tolerance for any substitution mutations in this region. This could possibly be due to a critical role for the C-terminal domain in Smpd1 dimerization (Zou et al., 1989).

Figure 3. smpd1 mutant larvae die shortly after hatching. Populations of homozygous and heterozygous late-stage embryos and early 1st-instar larvae sorted using the Biosorter. Heterozygotes can be identified using a fluorescent GFP marker. Heterozygote control, or homozygote larvae mutant for a small C-terminal deletion (∆24), or null smpd1 allele. 1st-instar smpd1 homozygous mutant larvae show widespread signs of cell death, or exhibit growth and tracheal development defects.
Taking a new approach
None of the smpd1 alleles we generated in the fly survived long enough to use in a high-throughput drug screen, so we have decided to take an alternative approach. We designed rescue constructs to express Drosophila smpd1, Homo sapiens (Hs) SMPD1, or Hs SMPD1 carrying the patient mutation R496L. We plan to use the GAL/UAS system to ubiquitously express these constructs in smpd1 null mutant flies. This will allow us to determine to what extent human SMPD1 can rescue the lethality we observe during late embryo/early larval stages. We can also determine if any observed rescue is reduced or eliminated when the patient allele is expressed. Finally, we can explore the possibility of bypassing an early requirement for smpd1 using conditional early expression that turns off after embryogenesis.
References
Levran O1, Desnick RJ, Schuchman EH (1991). Niemann-Pick type B disease. Identification of a single codon deletion in the acid sphingomyelinase gene and genotype/phenotype correlations in type A and B patients. J Clin Invest. 88(3):806-10.
Renault AD, Starz-Gaiano M, Lehmann R (2002). Metabolism of sphingosine 1-phosphate and lysophosphatidic acid: a genome wide analysis of gene expression in Drosophila. Mech Dev. 119 Suppl 1:S293-301.
Zhou Y-F, Metcalf MC, Garman SC, Edmunds T, Qiu H, and Weib RR (2016.) Human acid sphingomyelinase structures provide insight to molecular basis of Niemann–Pick disease. Nat Commun. 7:13082.
Zou L, Kojima N, Kito M, Yagi K (1989). Purification to homogeneity of human placental acid sphingomyelinase. Biotechnol Appl Biochem 11:217–225
